Research
topics
My
research interest is in the development of statistical machine learning methods
for analyzing real-world data. My previous research mainly focused on bio/chem data, but my current/future research is not limited to
these areas. If you are interested in working with me, contact me by the
following mail address.
saigo@inf.kyushu-u.ac.jp
Topic
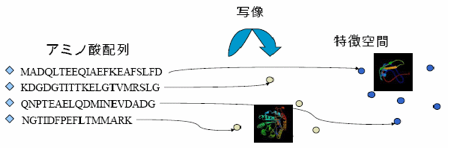
1: Prediction of protein functions
All
the living things including humans consists of proteins, and all the proteins consist
of amino acid sequences. In the era of Next Gen Sequencing, there are many
amino acid sequences available in the databases, but many of them are left
annotated.
In
my research, I have developed a kernel function that can capture similarity
among amino acid sequences based on well studied amino
acid substitution matrix and sequence alignment.
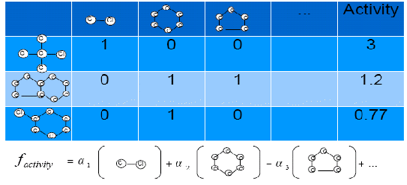
Topic
2: QSAR (Quantitative Structure Activity Relationship)
QSAR
(a.k.a. ligand based virtual screening) is one of steps in developing drugs in
pharmaceutical companies. In order to decrease the number of candidates
subjected to bio/chem experiments, computational methods
makes an important role.
In
my research, I am aiming at developing prediction methods that can not only equipped
with high prediction accuracy, but also equipped with interpretability to
humans. Since in drug development processes, it is important to work with bio/chem scientists, and convince them with explainable
prediction model.
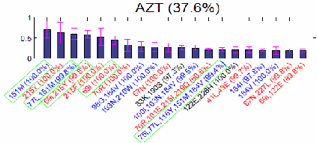
Topic
3: HIV drug resistance prediction
The
latest treatment of HIV/AIDS reads the sequence of HIV viruses in a patient. Each
virus has a different genetic information, and therefore behave differently
against the given drugs. Therefore it is important to
know the genetic information of viruses, then predict the amount of drug resistance
prior to selection of a therapy.
In
my research, I have developed a prediction method that can consider multiple
mutations in the amino acid sequence of a HIV-1 virus. Consideration of
multiple mutation turned out to be able to model the increase of drug
resistance by the accumulation of mutations around the active site of HIV-1
protein.